
A new method to analyze both RNA and proteins

Abstract
It is difficult analyze both RNA and protein expression in a single cell, because these two molecules have different chemical structures. In this article is shown a new technique, which allows the analysis of both molecular types in a single series of reactions using the C1 system (Fluidigm Corp.), by proximity extension assay and retrotranscription. In this way both proteins and RNA are decoded as a DNA signal.
In 2016 a research team born from the collaboration between Fluidigm Corp. and Massachusetts Institute of Technology published a research study in which a new technique to simultaneously analyze both RNA and protein expression is shown (1).
They assay to use an alternative technique to fluorescentce activated cell sortiung (FACS). FACS is a flow cytometry technique that allows to distinguish different types of cells present in a heterogeneous mixture. The distinction is made on the basis of the different characteristics of fluorescence and light diffusion.
Different fluorescent molecules are linked to the specific antibody for a protein present on the cell surface, in the nucleus and in the cytosol.
The cells are then passed through a laser beam with which they assume positive or negative charge, depending on whether or not they have the antibody bound on the surface.
Then they pass into an electric field that separates them. The cells are collected in different containers according to the type of antibody and fluorescent molecule bound on the surface. Homogeneous populations are thus obtained.
The relationship between RNA and protein expression is a feature still under study in biology, because there are long-lasting RNAs that give rise to proteins with short half-life. To get around this problem, scientists try to analyze the evolution of RNA and protein expression over time, in order to evaluate the variation of the two molecular species in different time phases.
To carry out these analyzes, the research team used a technique, still under development, that combines the retrotranscription and Proximity Extension Assay (PEA) (2) in a single series of reactions to convert the protein and RNA information into DNA, which is more stable and easier to amplify using qPCR.
To be able to analyze each cell individually, they used the Fluidmig C1 system, designed by the company in which some of the scientist work. The C1 system allows automated workflow, because reagents are provided to the machine, reducing contaminations. The C1 system also allows to create custom workflows in order to conduct different type of analyses on 96 single wells, like whole transcriptome analysis, gene expression profiling and gene regulation. Inside each well a single cell is placed in solution. To avoid mixing of the cellular content, each cell was lysed using a buffer containing the PEA probes.
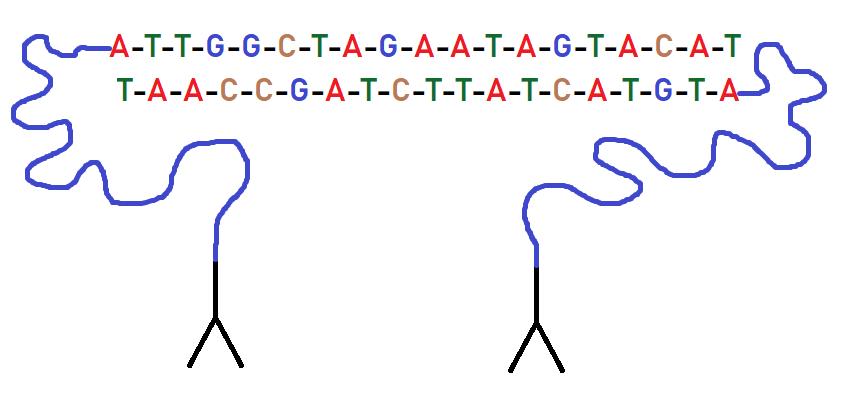
Figura 1 : PEA probes: they present a ssDNA complementary oligonucleotides.
Then the cells were incubated to allow the probes to connect with their protein target. They then performed the transcription coupled with DNA polymerization. This step is crucial for encoding protein and RNA information into specific DNA molecules. To analyze the abundance of proteins and RNA they used quantitative PCR (qPCR). In particular, a universal primer was used to detect all the molecules generated by the PEA probes. The cDNAs were amplified using gene specific primers in order to analyze only a limited number of RNAs and not all RNAs in the cell. They used PEA probes obtained in previous work, because usually they are intended for plasma analysis, but in this case is important that probes also reacted with intracellular targets
At this point they had to analyze the abundance of the original molecules in the cells, and they used high-throughput qPRC.
The experiment was conducted using MCF-7, a cell lineage derived from human breast adenocarcinoma treated with phorbol-12-miristate-13-acetate (PMA), an inductor of protein kinase C involved in apoptosis in MCF-7. The process was conducted on MCF-7 untreated, treated for 24h, and 48h, in order to evaluate the production and expression of a limited set of genes through time.
The research group studied a set of genes involved in apoptosis basing their decision on previous literature. In fact, has been shown that PMA activates protein kinase C signaling, that inhibit cell’s growth and induce apoptosis in MCF-7 (3). In particular it activates CASP 8, a master gene of a series of downstream genes that includes CCNE1, CDKN1B, EGFR, and RB1 (4-6).
The authors used this workflow to study expression of proteins and RNA related to cell apoptosis, obtaining some interesting results. They observed stimulation-induced changes in the seed network’s member, like cell cycle controllers and other intracellular and extracellular regulation genes.
Another annotation is about MET gene: they observed two melting-temperatures for the MET qPCR assay. This indicates the presence of two splice variants due to the primers they created. The preamplicon they designed were specific for exons 9 and 10, creating an amplicon that potentially spanned intron 9.
The article is basically technical. It proposes a new way to conduct multiplexed analyses in an interesting way, but we would underline the important effort given by Fluidigm Corp, and we think in this case how it is fundamental that independent research group tests the methods.
We found another critical issue, the material loss due to sample transfer and the complicated workflow; both problems could be bypassed using a custom automated machine. Using machines could help in several ways, maybe increasing the reliability of the system, which appear to be already very useful.
Moreover, we think that studying correlations between RNA and proteins in a large scale could not drive to a general deeper knowledge. We think that could be more useful using this method to investigate one RNA, or at least a smaller group of RNAs involved in a single signal cascade. If there is a common rule involving RNAs and proteins regulation, it could be found only analyzing single cases.
In conclusion although these problems we shown, we think that the technique is very interesting; the idea of transport both protein and RNA information in DNA molecule has been illuminating
References
- Genshaft, A.S., Li, S., Gallant, C.J. et al.Multiplexed, targeted profiling of single-cell proteomes and transcriptomes in a single reaction. Genome Biol 17, 188 (2016).
- Fredriksson S, Gullberg M, Jarvius J, Olsson C, Pietras K, Gústafsdóttir SM, et al. Protein detection using proximity-dependent DNA ligation assays. Nat Biotechnol. 2002;20:473–7.
- Oskoueian E, Abdullah N, Ahmad S. Phorbol esters from jatropha meal triggered apoptosis, activated PKC-δ, caspase-3 proteins and down-regulated the proto-oncogenes in MCF-7 and HeLa cancer cell lines. Molecules. 2012;17:10816–30
- He YY, Huang JL, Gentry JB, Chignell CF. Epidermal growth factor receptor down-regulation induced by UVA in human keratinocytes does not require the receptor kinase activity. J Biol Chem. 2003;278:42457–65
- Falk M, Ussat S, Reiling N, Wesch D, Kabelitz D, Adam-Klages S. Caspase inhibition blocks human T cell proliferation by suppressing appropriate regulation of IL-2, CD25, and cell cycle-associated proteins. J Immunol. 2004;173:5077–85.
- Maggio SC, Rosato RR, Kramer LB, Dai Y, Rahmani M, Paik DS, et al. The histone deacetylase inhibitor MS-275 interacts synergistically with fludarabine to induce apoptosis in human leukemia cells. Cancer Res. 2004;64:2590–600.