
DNA sequence and shape signatures collaborate in selecting different homeodomain transcription factor complexes

Figure 1 – Different TFs complexes bind to the DNA recognizing both its sequence and shape signatures. Moreover HthFL-Exd-Hox can have two configurations with different Hth orientation. [Image created with BioRender.com]
Abstract
Transcription factors (TF) can form various complexes, combining in different ways. However, understanding which complex a specific DNA sequence binds in vivo is still a complex and unsolved problem. Kribelbauer et al. showed that using SELEX-seq to find the DNA motifs bound and inferring DNA shape from them, it’s possible to assess the binding mechanisms of different complexes. Readout of the minor groove width is shown to play an important role in the interaction. Mutants with impairment in shape readout mechanisms can then be made, to analyze the role of the interaction in the binding in vitro and also in vivo
Discussion
In the past, it was thought that TFs bind to the DNA recognizing a specific motif, by contacting the major groove with the formation of hydrogen bonds. However it was proven that TFs binding is also affected by DNA structural features, such as DNA 3D structure, with a mechanism called ‘shape readout’ typically driven by electrostatic interactions between negatively charged amino acidic side chains and DNA minor groove [1]. The narrower the minor groove is, the stronger these interactions are. However, the two different types of interaction are difficult to distinguish, because the shape of the DNA is a consequence of its sequence [2].
Accurate prediction of how TFs complexes recognize and bind in vivo to a specific DNA sequence is still an unsolved problem, also due to the techniques used. Currently, the standard to analyze in vivo binding of TFs is chromatin immunoprecipitation sequencing (ChIP-seq), which however presents some limitations. In fact with this technique, it’s impossible to observe which TFs complex bind a specific sequence. In order to solve the problem, Kribelbauer et al. [3] used high throughput in vitro binding data together with DNA shape signatures to infer DNA shape readout mechanisms (Figure 1). Mutant TFs with impairment in these shape readout mechanisms were then designed. By comparing wt and mutant TFs’ behavior in vivo, authors obtained detailed information on how different complexes interact with the DNA and control gene expression.
As a model to test their approach, researchers used a system of three homeodomain TFs from Drosophila Melanogaster: Hox, Extradenticle (Exd), and Homotorax (Hth). All three are involved in embryonic development. Two major isoforms of Hth exist. The full lenght one (HthFL), which contains a homeodomain and so can bind to the DNA. Instead the other one, Hth Homotorax-Meis (HthHM), only contains the HM domain and so cannot bind to the DNA, but still allows nuclear localization of Exd, that happens thanks to Exd-Hth interaction through the HM domain. These three TFs form various complexes with different compositions: Hth monomer (Figure 1A), HthFL homodimer (Figure 1B), HthFL-Exd heterodimer (Figure 1C), HthHM-Exd-Hox (Figure 1D), and HthFL-Exd-Hox (Figure 1E).
In vitro analysis
First of all, Kribelbauer et al. identified the binding sequences of the different complexes using systematic evolution of ligands by exponential enrichment sequencing (SELEX-seq) [4]. In this technique, first of all DNA libraries are made and TFs of interest are added to them. Unbound sequences are discarded, while bound ones are recovered and amplified with PCR. Multiple rounds of enrichment can be performed, to obtain sequences with higher binding affinity. To see binding of the dimeric complexes, a 16 bp random region library was used. Whereas to see the binding of trimers, two different 21 bp random regions libraries were made, both containing a fixed Hth binding site: Hth forward library and Hth reverse library. Results were summarized in position specific affinity matrices (PSAM) that showed the binding motif of each complex.
To assess the preferred configuration of the trimeric complex with full length Hth, free energy of the relative position and orientation of Hth and Exd-Hox subcomplex was considered. Using PSAM, a preference for the conformation with Exd facing Hth emerged (Figure 1E). Moreover, researchers also analyzed the length of the spacer between Hth and Exd. Using Hth reverse library, a preference for longer spacers, 3-10 bp, was observed. While in the Hth forward one, spacers shorter than 4 bp were preferred. This is due to Hth N-terminal being closer to Exd (Figure 1F) in the reverse library compared to the forward one.
Afterwards the authors tested if the spacer played a role in binding affinity of the complex. In the PSAM, an enrichment for AT-rich sequences was found, suggesting that shape readout of the minor groove might be involved. In fact, AT-rich sequences tend to show narrower minor grooves. To prove their hypothesis, researchers made a model that was able to predict width of the minor groove, using SELEX-obtained sequences as input [5]. Firstly, the authors observed a narrowing of minor groove width (MGW) minima with the increase of the sequences’ binding affinity. Secondly, they also found two MGW minima close to the spacer, one selected by Hth and the other by Exd (Figure 2). In addition, Hox selected two MGW minima adjacent to its binding site.
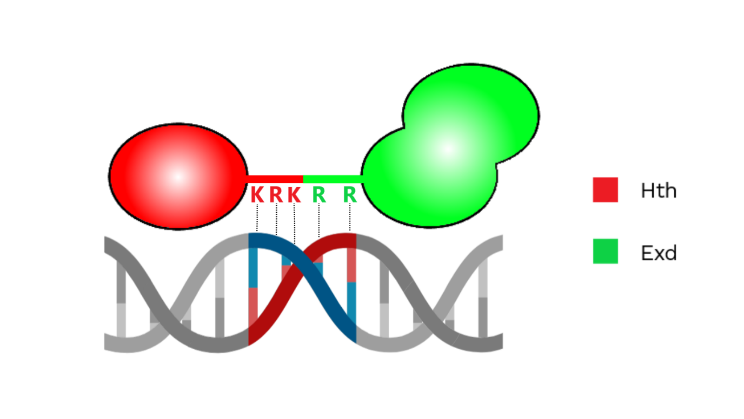
Figure 2 – Representation of Hth and Exd interacting with the minor groove with positively charged amino acids. [Image created with BioRender.com]
To prove that Hth and Exd exploit MGW readout mechanisms, mutations in positively charged amino acids were made. In particular, two arginines were mutated in alanines in Exd. While in Hth, mutations of two lysines and one arginine to alanines were performed. A loss in selection of at least one MGW minima with both mutants was observed. Researchers saw that mutant Exd also caused the loss of selection for one of the Hox minima, so they used it to assess if shape readout of the minor groove was required for all Exd-containing complexes. Hox-containing complexes were more affected by the mutation in particular the trimer with HthHM completely lost the ability to bind to the DNA.
In vivo analysis
By using whole genome ChIP-seq in wing imaginal discs of Drosophilas Melanogaster with either wt or mutant Exd, the authors proved that the binding of the different TF complexes recapitulated the patterns seen in vitro. Researchers wanted to understand the cause of Exd mutation lethality, so they focused on ChIP-seq peaks lost when Exd was mutated.
These showed high affinity for HthHM-Exd-Hox according to SELEX-seq derived PSAM and other models [6]. Then researchers thought that loss of binding affinity together with sequence affinity obtained with in vitro SELEX might allow to assign to each Exd ChIP-seq peak a particular homeodomain complex. So clustering of the different peaks was performed, using information from 3 ChIP-seq and 3 predicted affinity scores along with loss of binding upon Exd mutation. Seven of the eight clusters identified were assigned to a specific type of complex with low or high affinity for its binding site, while the last cluster, called motifless, lacked a clear motif. Moreover, chromatin accessibility, assessed with ATAC-seq, was shown to be correlated with complex composition and configuration, suggesting that different complexes can differently affect DNA accessibility and gene expression.
The authors then used mutant Exd to identify the gene network controlled by Hox-containing complexes, since this mutant impacted their binding the most. The authors performed RNA-seq in wing imaginal discs, finding 392 upregulated genes and 322 downregulated ones. With in situ chromatin capture [7], the correlation between change in gene expression and promoter-Exd peaks cumulative contact frequency was analyzed. A correlation was found in upregulated genes but not in downregulated ones. So researchers suggested that Exd containing complexes could recognize their target sites by forming transcriptional hubs (Figure 3), in which many TFs from various enhancers and promoters are concentrated, forming a 3D microenvironment. In order to prove their hypothesis, whole genome in situ chromatin capture was performed. More frequent colocalization of Exd containing complexes compared to random ones was observed.
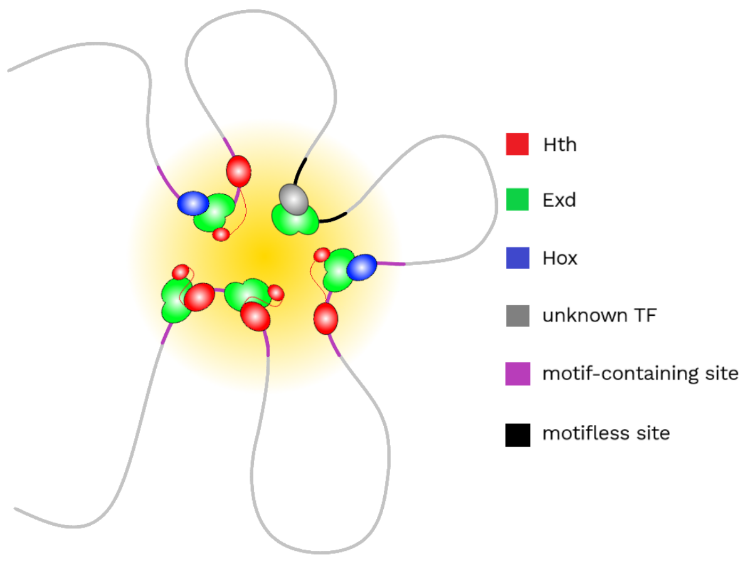
Figure 3 – Representation of a transcriptional hub, in which motifless sites are colocalized with motif-containing sites.
Lastly, gene ontology (GO) analysis of the most frequently contacted promoters by each peak was performed. When considering each different complex separately, researchers found an enrichment in various functions, that were missed when considering all Exd-peaks together. Many of these functions were related to neuronal categories, such as chemotaxis, axon guidance, cell projection and cell morphogenesis. In addition, by analyzing gene ontology of motifless sites, a similar enrichment in functions as when considering all Exd-peaks was observed. Researchers had also seen a large variability in loss of binding of motifless sites with mutant Exd, similar to the loss observed in motif containing sites. So they hypothesized that motifless sites are occupied to a certain degree by Exd-containing complexes and are in close proximity or even in contact with sites containing a motif.
Conclusions
In conclusion, with the approach illustrated here, it was possible to understand the sequences bound by different TFs complexes in vitro and the in vivo data obtained well recapitulated the in vitro findings. Moreover, thanks to the analysis made on in vivo binding data obtained, it was also possible to reveal new complex-specific functions by performing gene ontology analysis of the promoters contacted by one particular complex’s peaks. Researchers suggested that these new GO functions found may help in explaining seemingly contradicting roles of Hox in formation of the nervous system and cancer. In addition, Kribelbauer et al. also suggested that mutations similar to those analyzed in this article, may play a role in human CAKUT (congenital anomalies of the kidney and urinary tract) syndrome. In fact, four Exd orthologs are present in humans: Pbx1-4. These TFs are very conserved and are critical for viability in mice. CAKUT syndrome was correlated with de novo mutations in Pbx1 [8], which the authors speculated could cause impairment in shape readout mechanisms of the minor groove, thus leading to the pathology.
We believe that the approach proposed is well-suited to address such a complex issue as the one considered in this paper. In the future, it would be interesting to further study the motifless cluster. It had already been shown that low affinity binding sites have a role in the specificity of binding of Exd-Hox containing complexes [9] and the authors hypothesized that similar sites, in this paper called motifless sites, could be in contact with complexes containing a motif. So, focusing on the peaks of the motifless cluster, in the future it may be interesting to test by which TFs complexes they are bound by and how they are recruited as a part of Exd-containing complexes transcriptional hubs. Moreover, it could also be needed to perform similar experiments using this technique with other TFs, with lower binding affinity or that form bigger complexes, to better understand the extent of this approach. Lastly, analysis of in vitro binding data and DNA shape signatures could, in the future, be also used to better understand the cause of some pathologies, such as CAKUT syndrome.
References
- Slattery M., Zhou T., Yang L., Dantas Machado A.C., Gordân R., Rohs R., 2014.Absence of a simple code: how transcription factors read the genome. Trends in Biochemical Sciences, 39(9):381-399.
- Abe N., Dror I., Yang L, Slattery M., Zhou T., Bussemaker H.J., Rohs R., Mann R.S., 2015. Deconvolving the recognition of DNA shape from sequence. Cell, 161(2):307-318.
- Kribelbauer J.F., Loker R.E., Feng S., Rastogi C., Abe N., Rube H.T., Bussemaker H.J., Mann R.S., Context-Dependent Gene Regulation by Homeodomain Transcription Factor Complexes Revealed by Shape-Readout Deficient Proteins. Molecular Cell, 78(1):152-167.e11.
- Riley T.R., Slattery M., Abe N., Rastogi C., Liu D., Mann R.S., Bussemaker H.J., 2014. SELEX-seq: a method for characterizing the complete repertoire of binding site preferences for transcription factor complexes. Methods in Molecular Biology, 1196:255-278.
- Zhou T., Yang L., Lu Y., Dror I., Dantas Machado A.C., Ghane T., Di Felice R., Rohs R., 2013. DNAshape: a method for the high-throughput prediction of DNA structural features on a genomic scale. Nucleic Acids Research, 41:W56-62.
- Rastogi C., Rube H.T., Kribelbauer J.F., Crocker J., Loker R.E., Martini G.D. et al., 2018. Accurate and sensitive quantification of protein-DNA binding affinity. Proceedings of the National Academy of Sciences of the United States of America, 115(16):E3692-E3701.
- Rao S.S., Huntley M.H., Durand N.C., Stamenova E.K., Bochkov I.D., Robinson J.T. et al., 2014. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell, 159(7):1665-1680.
- Slavotinek A., Risolino M., Losa M., Cho M.T., Monaghan K.G., Schneidman-Duhovny D. et al., 2017. De novo, deleterious sequence variants that alter the transcriptional activity of the homeoprotein PBX1 are associated with intellectual disability and pleiotropic developmental defects. Human Molecular Genetics, 26(24):4849-4860.
- Crocker J., Abe N., Rinaldi L., McGregor A.P., Frankel N., Wang S. et al., 2015. Low affinity binding site clusters confer hox specificity and regulatory robustness. Cell, 160(1-2):191-203.