
Genome-Wide DNA Methylation Profiling Reveals Epigenetic Changes in the Rat Nucleus Accumbens Associated with Cross-Generational Effects of Adolescent THC Exposure

In an article of 2015 the research team led by Yasmin Hurd in New York using Enhanced Reduced Representation Bisulfite Sequencing interrogated the DNA methylation of the nucleus accumbens of rats and identified 1027 differentially methylated regions (DMRs) associated with parental THC exposure in adults [1]. The Δ9-tetrahydrocannabinol (THC), is a fundamental psychoactive principle of Cannabis Sativa: this is the soft drug most consumed in the world with a spread that increases year after year. It is an important object of study since the effects of this drug’s chronic intake are not well understood both in individuals who use it, or in subsequent generations, especially considering its wide use for recreational and medical purposes [2]. In a first experiment of the same research group the correlation between THC intake and behavioral alterations in the progeny of rats exposed to the drug has been demonstrated; these alterations involved the increase of stereotypical behaviors, compulsive heroin research and altered synaptic plasticity due to the alteration of mRNA levels of genes responsible for this function and for synthesis of cannabinoid receptors in Nucleus Accumbens cells (NAc) [3].
This part of the brain, located in the striatum, has been examined as the seat of the cognitive processes of motor function related to the reward or positive reinforcement of the action [4]. Following these initial results Corey T Watson et al. considered necessary to examine the levels of alteration of methylations in these cells. DNA methylation is an epigenetic modification often related to altered levels of gene expression, normal brain development, environmental situations and is often passed on to subsequent generations through genomic imprinting [5]. The authors used a Cross-Generational THC Animal Model, from which 32 specimens were derived and NAc tissue from their brains was dissected obtaining:
– 16 “THC” specimens, rats whose parents had been exposed to the narcotic substance;
– 16 “VEH” specimens, rats for control whose parents had been exposed to the missing “vehicle” of the substance.
They evaluated the genome-wide methylation level using the Enhanced reduced Representation Bisulfite Sequencing (ERRBS) technique [6]. This allowed them to analyse the differentially methylated regions (DMRs) between the THC and VEH samples, in the 20 autosomes. They found 406 hypermethylated and 621hypomethylated DMRs in the offspring with parents exposed to THC, including 5611 CpGs. The DMRs identified had percentages of methylation ranging from 3.6% to 11.3%. Although the modifications are modest, these have been detected especially within the gene bodies and in the promoter zones.
This is a very important fact because the methylations in promoter regions generally inactivate the transcription of the gene downstream of the same, whereas methylations in genes body result in an hyperexpression of the gene itself or alterations of its splicing pattern, according to the different levels of mRNA expression in NAc cells highlighted in the first experiment. The research group has undertaken to analyze not only the methylation levels at the single gene level but also to compare them according to the protein-protein interactions that occur between the proteins encoded by the same genes.
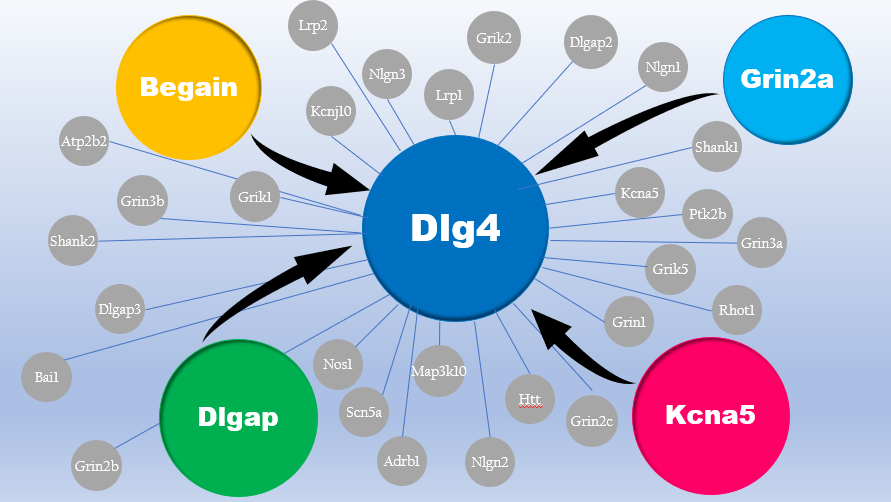
An example is the Grin2a gene, a regulator gene of synaptic plasticity, which has a hypomethylation level of about 2.5% lower, whose protein interacts in the Dlg4 Pathway with other proteins whose genes (Dlgap2, Kcna5, Begain and Dlg4) present themselves different methylation levels [6]. However, it is important to underline that the ERBBS technique enriches the CpG regions present above all at the promoters level, so it would be necessary to carry out these analyzes again using a whole genome sequencing after bisulfite treatment.
In conclusion, this research provided the first dataset on intergenerational epigenomic alterations associated with THC exposure in rat NAc, a brain area related to addictions. These results open the way for new studies that can focus on:
- correlation between epigenetic alterations and transcriptome;
- molecular mechanisms through which exposure to THC causes epigenthic alterations at the level of NAc cells;
- understanding of the molecular mechanisms underlying the heritability of epigenetic alterations in relation to parents’ THC exposure;
- differences in methylation and effects of THC exposure dependent on sex.
References
- Corey T Watson, Henrietta Szutorisz, Paras Garg, Qammarah Martin, Joseph A Landry, Andrew J Sharp and Yasmin L Hurd. Genome-Wide DNA Methylation Profiling Reveals Epigenetic Changes in the Rat Nucleus Accumbens Associated With Cross-Generational Effects of Adolescent THC Exposure, Neuropsychopharmacology, 2015
- Henrietta Szutorisz, Yasmin L. Hurd. High times for cannabis: Epigenetic imprint and its legacy on brain and behavior, Neuroscience and Biobehavioral Reviews, 2018
- Szutorisz H, DiNieri JA, Sweet E, Egervari G, Michaelides M, Carter JM et al. Parental THC exposure leads to compulsive heroin-seeking and altered striatal synaptic plasticity in the subsequent generation. Neuropsychopharmacology, 2014
- Sanjay Salgado, Michael G. Kaplitt. The Nucleus Accumbens: A Comprehensive Review, Stereotact Funct Neurosurg, 2015
- Gutierrez-Arcelus M, Ongen H, Lappalainen T, Montgomery SB, Buil A, Yurovsky A et al. Tissue-specific effects of genetic and epigenetic variation on gene regulation and splicing. PloS Genet. 2015
- Alexander Meissner, Andreas Gnirke, George W. Bell, Bernard Ramsahoye, Eric S. Lander and Rudolf Jaenisch. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005