
scCAT-seq: a breakthrough into multi-omics layers investigation at single-cell level
The necessity to further comprehend the heterogeneity between single cells, as well as the ability to study the cell lineage trees, become clear with the progress in studying of the epigenome, genome, and transcriptome. A major obstacle in understanding the individual differences among single cells was the difficulty to integrate -omics data coming from different bulk profiles. With this revolutionary technique, able to investigate at the same time, and in the same cell, the epigenome and the transcriptome layers, deeper levels of information can be reached. Also, it will possible to study all the cases in which there is extraordinarily low amount of sample, such as rare or circulating tumour cells.
The regulatory heterogeneity between different cells become clear with the developing, in the last few years, of an increasing number of techniques able to efficiently depict the complexity of single cells in terms of epigenetic, genetic and transcriptional regulation [1]. While several omics techniques have already been established at the single cell level, and have been successfully applied to analyse complex cell population such as tumorigenesis, cellular reprogramming and developmental processes [2-4], there is still lack of methods able to experimentally integrate the multi-omics layers of information at the single-cell level, thus allowing to fully understand how the regulation works. This article fits in this scenario, causing a dramatic breakthrough in understanding the regulation scenario of the multi-omics layers in single cells.
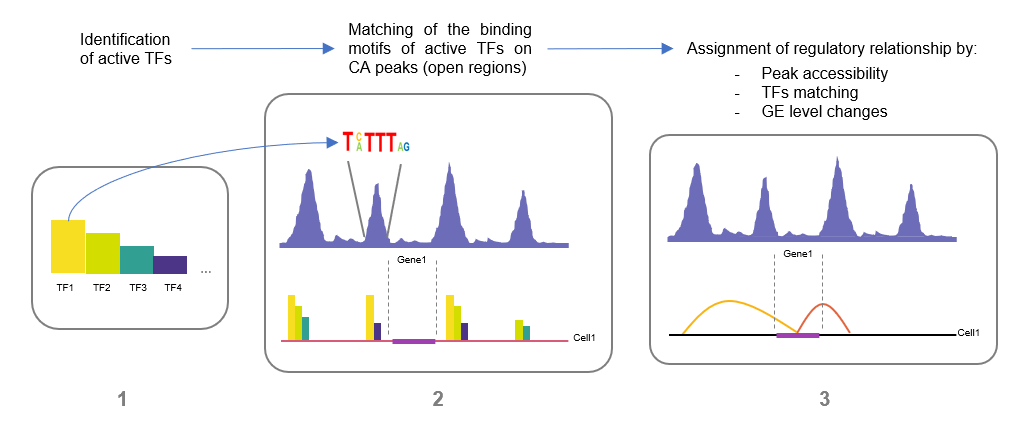
Fig.1
In figure it is shown the three-points strategy applied to analyse the gene expression (GE) data and the chromatin accessibility (CA) data to infer the regulatory relationship between CREs regions and the putative gene target expression.
IN a recent work Liu and collaborators [5] illustrate a novel technique called scCAT-seq (single cell chromatin accessibility and transcriptome sequencing). The strength of the scCAT-seq is the ability to investigate, at single cell-level, both the epigenome state (e.g. chromatin accessibility) and the transcriptome state. The first part of the article is focused on the validation of this technique, both from a technical point of view and from the functional one. Regarding the technical validation, they tested and verified the ability of the scCAT-seq in efficiently depicting the chromatin accessibility state and the gene expression level compared to the previous techniques. From the functional point of view, they proved that this technique was effectively able to describe the transcriptional state of the cells, both when investigating the structure of the CREs (cis-regulatory elements, such as histone modifications) and the binding of the trans-factors (such as transcription factors). Subsequently they applied the scCAT-seq to identify the regulatory relationships between the accessible chromatin CREs sites and the putative genes target expression level in each cell. They used the following strategy. First, they identified the active TFs for every cell, based on the co-expression of the TFs and their target genes across cells. Second, they identify the active accessible regions by matching the binding motifs of the active TFs previously found to the accessible chromatin regions. Next, they assigned the regulatory relationships if the presence of the active accessible region matched with the active TFs binding motifs was associated with a significant change in the expression level of the gene target. The regulatory relationships detected with this strategy were subsequently validated with the ChIA-PET. The result was a binary matrix, with the single cells as columns and the regulatory relationships as rows, and where the entries show the presence or not of that particular regulatory relationship in each cell. This matrix was then decomposed by a clustering analysis, the NMF (non-negative matrix factorization package), to rearrange the matrix based on the presence or not of the regulatory relationship. The resulting heatmap based on the active regulatory relationship was able to clearly distinguish all the cells in three different population, equivalent to the three different cell lines investigated by the authors (K562, HeLa-S3, and HTC116).
The conclusive part of the article is an experiment on human pre-implantation embryos, which aim to seek whether this technique can distinguish between complex cell populations. During the development, the human pre-implantation embryos undergoes through spectacular changes both in the chromatin accessibility state and in the transcriptional activity. To test the ability of the scCAT-seq in taking a faithful picture of the regulatory relationships in this sensitive phase, they performed the previous described steps, finding around 100 k regulatory relationship and the subsequently separating all the cells in two distinct cell populations, one belonging to the blastocyst stage (43 cells) and the other to the morula stage (29 cells). Next, they analysed the TFs accessibility and their relative gene expression level to identify the key regulatory TFs characterizing the two different stages. This analysis succeeded in distinguishing again the two cell populations based on the pluripotency markers, typical of the morula stage, and the trophectoderm markers, typical of the blastocyst stage, proving that this integrated approach could faithfully identify distinct subtypes from the same origin.
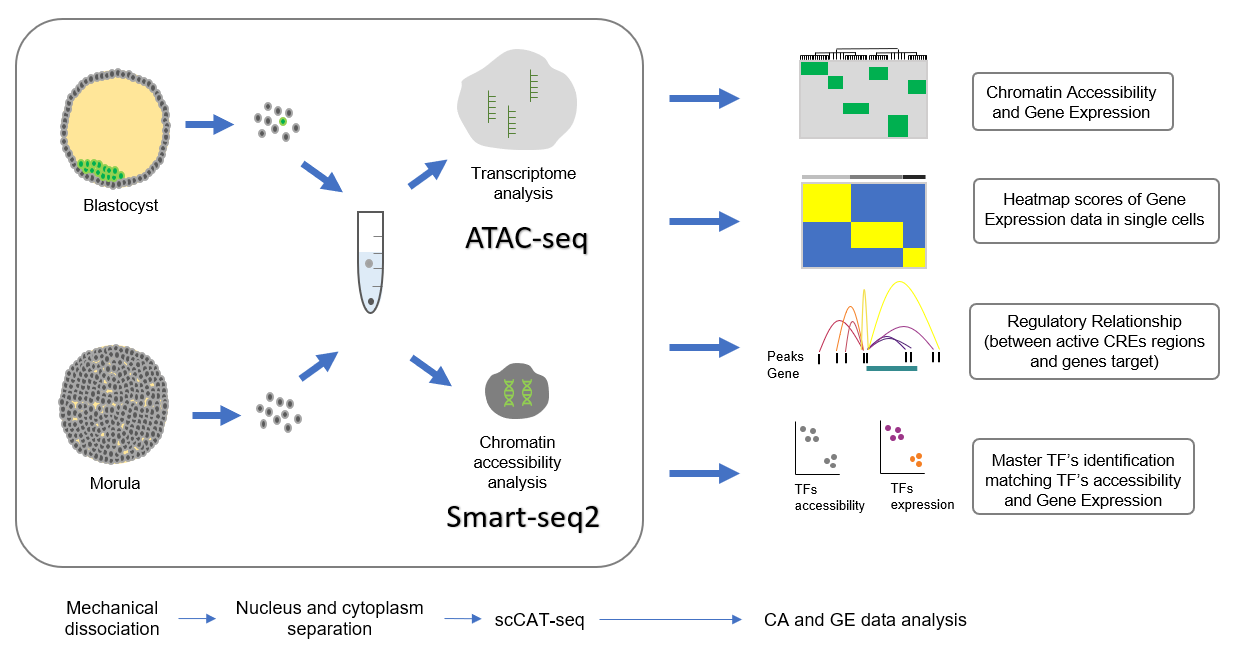
Fig.2
In figure it is shown the workflow of the experiment performed by the authors on the human pre-implantation embryos, and the information they could obtain analysing the CA and GE data.
The scCAT-seq combined with the author’s approach allowed a high resolution epigenomic and transcriptomic portraits of individual cells, the discovery of functionally relevant regulatory relationship and the identification of master TFs in complex cell population, and finally the characterization of a transition state, such us the human embryos development. Therefore, this technique can be considered an impressive breakthrough in the multi-omics layers investigation at single-cell resolution, providing a view of countless possible application. Among other valuable applications, there is the analysis of rare or circulating tumour cells and the study of tumour heterogeneity and microevolution (cell lineage reconstruction of cancer to elucidate its development), but also the characterization of several differentiation processes, that could lead to the manipulation of the cell fate-commitment. Finally, it will also be a precious help whenever it would be necessary to perform any kind of analysis on a significant small amount of sample.
To conclude, the authors were extremely dutiful in sewing a small masterpiece, trimmed at 360 degrees, from the technique itself to the strategy needed to read the resulting raw data, and finally to the different experiments they performed on more complex models (from cell lines to the human embryos).
References
- Liu et al., Deconvolution of single-cell multi-omics layers reveals regulatory heterogeneity., Nature Communications, 10:470 (2019)
- Shapiro, E., Biezuner, T. & Linnarsson, S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat. Rev. Genet. 14, 618–630 (2013).
- Wen, L. & Tang, F. Reconstructing complex tissues from single-cell analyses. Cell 157, 771–773 (2014).
- Buenrostro, J. D. et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490 (2015).
- Buenrostro, J. D. et al. Integrated single-cell analysis maps the continuous regulatory landscape of human hematopoietic differentiation. Cell 173, 1535–1548 e1516 (2018).