
A new assay that allows to discriminate cellular identity from a heterogeneous population

Abstract
A new single-cell combinatorial indexing for methylation analysis (sci-MET) protocol has been developed. To test its efficiency 3,282 single-cell bisulfite sequencing libraries were produced with a reading alignment rate of 68 ± 8% achieved. Here are reported the results of the application of the sci-MET protocol able to recognize the identity of individual cells from a mixed cell population.
The complex relationship between the presence of a covalently linked methyl group in cytosine-guanine dinucleotides (CG) and non-CG (CH sites) and gene expression of the nearby gene has been known for years [1]. The increase interest in the cell methylation profile has made it possible to expand the studies connected to DNA methylation, allowing to discover the alterations also in pathological contexts such as cancer and identify the differences between the various cell types [2]. The specificity by cell type and the changes undergone during development make the methylation profile an interesting tool for cell identification starting from a complex structure such as a tissue, stimulating a single-cell approach [1].
For this purpose, Ryan M. Mulqueen and collaborators, in a publication in “Nature Biotechnology” in 2018 [3], proposed an assay that exploits single-cell combinatorial indexing and the whole genome bisulfite sequencing (WGBS), through which it is possible to probe the DNA methylation at base pair resolution. This assay has been called single-cell combinatorial indexing for methylation analysis (sci-MET).
Sci-Met workflow
This technique, initially, involves the isolation of cell nuclei and the modification of the DNA contained by depletion of the nucleosomes followed by the insertion of a first index through the use of transposases. The nuclei with the mono-indexed sequences, after being pooled and randomly redistributed equally in a certain number of wells, were subjected to treatment with the bisulfite with the insertion of a second index by PCR, thus generating a cell-specific barcode. The final step is represented by sequencing (Fig 1).
The cited protocol was also optimized: using transposons with cytosine-depleted indexes not to be influenced by bisulfite treatment and exploiting the crosslinking/SDS technique for nucleosome depletion. The latter made it possible to keep the collision rate lower (two nuclei of the same transposase barcode ending up in the same PCR well), compared to the lithium assisted nucleosome depletion (LAND) technique, ensuring nuclear integrity in line with what has been reported in previous studies in which this factor is described as the key hurdle to produce uniformly distributed sequence reads [4].
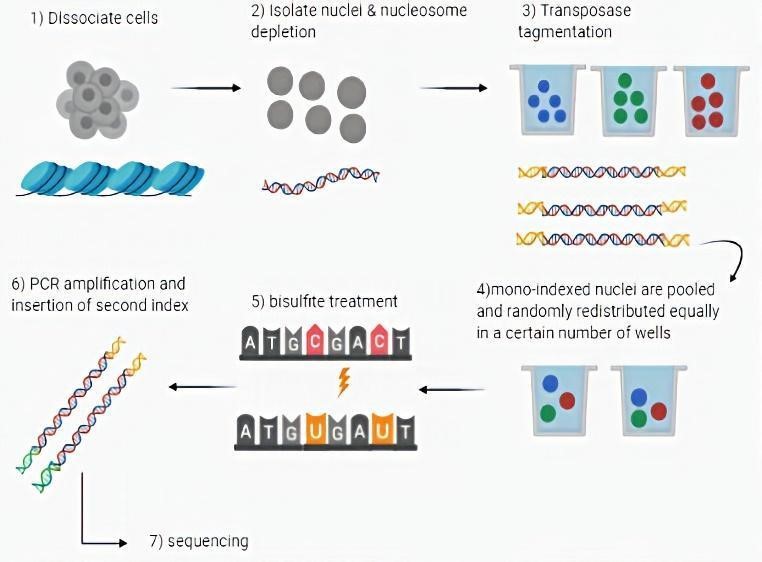
Figure 1 The sci-MET workflow
Application of the protocol and bioinformatics analysis
Through the application of the protocol on pure cell lines and on an artificial mixture of human B-lymphoblast cell lines (GM12878), primary inguinal fibroblasts (GM05756) and HEK 293 cells (for a total of 880 cells), followed by the alignment of the reads with the human genome, the researchers were able to characterize genome-wide methylation in 691 cells, achieving an efficiency peak of 78.5% and an average alignment of 68 ± 8%.
The experiment continued alongside the procedure described with a bioinformatics analysis, which consisted of the application of the non-negative Matrix Factorization (NMF) followed by t-distributed Stochastic Neighbor Embedding (t-SNE), which allows a better analysis of the data by a reduction in dimensionality, and by identifying clusters using density-based methods.
Finally, cluster methylation rates were correlated with publicly available WGBS data sets for the first 1,000 most variable regulatory regions. For each merged cluster, the two most correlated samples were of the same cell type or the most similar cell line.
Cell type discrimination in an in vivo model
To test the possibility of performing cell discrimination in an in vivo model, the previously described procedure was applied to a population of primary cortical cells from three different mice. After clustering, the coverage of the various clusters was aggregated and the percentage of methylation of specific cortically differentiated methylated regions (DMRs) was measured. This revealed a marked enrichment for each neuronal cluster within sets of excitatory and inhibitory DMRs and allowing classifications of clusters to be classified.
Strengths and limitations
Although a limitation of the technique consists in not being, at present, the one that allows the best coverage if compared with what reported in other studies [5], it is able to guarantee sufficient coverage per cell to identify the cell identity starting from a mixed population maintaining a good cost / efficiency ratio. However, it should be highlighted the possibility of increasing the efficiency of the technique through additional rounds of linear amplification, or by exploiting its scalability, expanding the number of indexes.
References
- Varley, K.E. et al. Genome Res. 23, 555–567 (2013).
- Smallwood, S.A. et al. Nat. Methods 11, 817–820 (2014).
- Mulqueen et al., Biotechnology 36, 428-431 (2018)
- Vitak, S.A. et al. Methods 14, 302–308 (2017).
- Clark, S.J. et al. Nat. Protoc. 12, 534–547 (2017).