
An innovative non-linear alignment algorithm to compare non-coding RNAs

Figure 1 – A visual explanation of k-mer counting.
Abstract
Long non coding RNAs (lncRNAs) have been recently demonstrated to regulate many biological processes. However, the understanding of their classification still remains challenging. A new approach, developed by Kirk and collaborators [1] may reveal homologies that previous algorithms fail to detect. Thanks to this novel tool it is now possible to infer the function of a lncRNA, providing the researchers a useful starting point for further functional characterization of the analyzed sequence.
LncRNAs are implied in a large number of genetic regulation processes[2], spanning from chromatin remodeling to regulation of the stability of mRNAs. The most recent studies have begun to uncover the mechanisms behind their biogenesis and how most of their functions depend on their localization within the cell and on the interactions they establish with proteins, RNA and/or DNA.
The most common computational approach to functional classification of biological elements is the alignment of the sequences of these elements to annotated sequences in online databases[3]. This allows to highlight the most conserved parts of the sequences, which are likely to give the sequence (DNA, Protein, RNA…) its function, and characterize it indirectly. The main issue with lncRNAs is the almost complete lack of linear sequence homology for most transcripts. Accordingly, discerning the function of one lncRNA does not yield insights into the functions of other lncRNAs, which leads to the necessity of direct studies to explore their biological properties [4].
The new algorithm that is presented in the paper, named SEEKR (Sequence Evaluation from K-mer Representation), tackles this problem directly by introducing non-linear sequence alignment using k-mer content as a proxy to infer the function based on similarity to other lncRNAs. K-mer is a short nucleotide sequence of variable length, between three and eight bases, where ‘k’ stands for the length of the motif (Fig.1). To achieve this, SEEKR performs a very simple series of steps: given a group of sequences and a value for k, i.e. the length of the k-mer, it counts every occurrence of each k-mer for all the analyzed lncRNA sequences. After normalizing the counts over the length of the RNA sequence, SEEKR creates a k-mer profile for each of them, which is the vector of the z-scores calculated through the counts. These z-scores are used to calculate the Pearson correlation value between each pair of lncRNAs: the resulting matrix contains a score between 1 and -1 for each sequence pair. The score indicates the strength of correlation: a score close to 1 or -1 indicates a significant positive or negative correlation, respectively; a score close to 0 means there is little to no correlation between the lncRNAs. Strong correlation of a novel lncRNA to a well-characterized one could guide the researcher to predict its function.
During validation of the algorithm, the authors tested and compared its ability to identify known sequence similarities between evolutionarily related lncRNAs, with two existing alignment algorithms (nhmmer [5] and Stretcher [6]). SEEKR ranks at second place, surpassing Stretcher while staying behind nhmmer. Following this, SEEKR is tested on human and mouse lncRNAs with unknown sequence homology to find out if it was able to detect new forms of similarities that were unseen by conventional algorithms. Firstly, they applied a hierarchical clustering and observed specific patterns. Secondly, they used a network-based approach (Louvain method) to partition the lncRNAs into communities based on their k-mer content. SEEKR was effectively able to group the sequences into 5 communities, which were further characterized.
LncRNA subcellular localization inferred from k-mer content similarities was investigated through available data from the ENCODE Project [7]. Each lncRNA expressed in HepG2 and K562 cells was sorted by nuclear ratio and divided in the communities identified in the previous test. Kirk et al. were able to find significant differences between communities 1 and 3, which were accordingly labelled as the most nuclear and cytoplasmic, respectively. This asymmetrical distribution was also reflected in the k-mer content, with 58% of 360 cytoplasmic k-mers and 93% of 27 nuclear k-mers enriched in the communities 3 and 1, respectively. Therefore, these results prove that SEEKR could predict the subcellular localization of a lncRNA from its k-mer content.
To test the ability of SEEKR to predict protein-RNA association, the authors employed a database of known RNA-protein interactions [7,8]. They developed two logistic regression models for each protein in the database and showed that the addition of lncRNA community assignments as a predictor improved the outcome. In particular, the full model significantly contributed to increase the log-likelihood of detecting RNA-protein associations as wells as precision and recall.
Protein binding is further explored to the extent of the degeneration of binding motifs. Different lncRNAs that bind the same protein possess sequence homology that is beyond the canonical binding motif, and this was measurable through k-mer content. By employing SEEKR. eCLIP data from the ENCODE Project revealed that often lncRNAs do not precisely interact with the proteins in the binding motif region. The k-mer content of 300-nt long windows around each motif match was analyzed and pairwise comparisons allowed to conclude that.
In addition to this, the most common k-mers inside interaction regions for each protein have been compared to binding motif determined in vitro. The results suggest two distinct patterns: for some proteins there is a strict homology between the binding motif and the most common k-mers, whereas a second group of proteins shows little to no homology between the most common k-mers and the binding motif. The authors speculate that the RNA-protein interactions of the first group are mainly determined by the density of these k-mers in the binding motifs, whereas for the second group, possibly unknown mechanisms involving sequence segments outside of the binding motif drive the formation of interactions.
While all the assays up to now aimed to determine the function of lncRNAs indirectly, using localization or the bound proteins as a means to understand their function, and most importantly have been fully in silico approaches on already available data, the following step is mixed, with the introduction of a new assay for lncRNA cloning in cells which tries to infer the function of a certain lncRNA directly. Kirk’s team wants to uncover whether k-mer content gives direct information for the prediction of the function of a lncRNA: to do so, they studied cis-repression by the XIST RNA, which is well-known to act as the master regulator of the X chromosome inactivation, providing dosage compensation in mammals. The choice was driven by the previous knowledge and the well-known inactivation mechanism [9].
The newly developed assay was called TETRIS (transposable element to test RNA’s effect on transcription in cis) and allows the use of the Tet-On gene expression system on a lncRNA sequence (Fig.2). The lncRNA is close to the reporter gene (Luciferase), which has a constitutive promoter, and is regulated by a doxycycline-induced promoter. Different modifications to XIST were tested: it ultimately resulted that all of the sequence elements necessary for repression were included in the first 2 kilobases (Xist-2kb) and that one of these is called repeat A (repA). The deletion of repA caused a significant but not complete de-repression of luciferase. Moreover, the results showed that the expression of repA alone did not reproduce the total amount of repression, implying that some other sequence element was also responsible for it.
Investigation of the k-mer content of Xist-2kb involved the creation of 6 artificial lncRNA sequences with increasing k-mer content similarity to Xist-2kb. When tested in vitro, these sequences caused luciferase repression, with more similar sequences having a stronger effect. The authors tested whether the 3 alignment algorithms could negative correlation between sequence similarity and luciferase signal. Only SEEKR was able to give meaningful results, with nhmmer not finding any similarity to Xist-2kb and Stretcher finding as little as 3% difference only between the first and the sixth sequence. The same analysis is repeated with other 33 lncRNA. Once again, only SEEKR was able to find a meaningful negative correlation between similarity to Xist-2kb and the level of luciferase. The authors conclude that k-mer content can be used to evaluate cis-repressive function of lncRNAs.
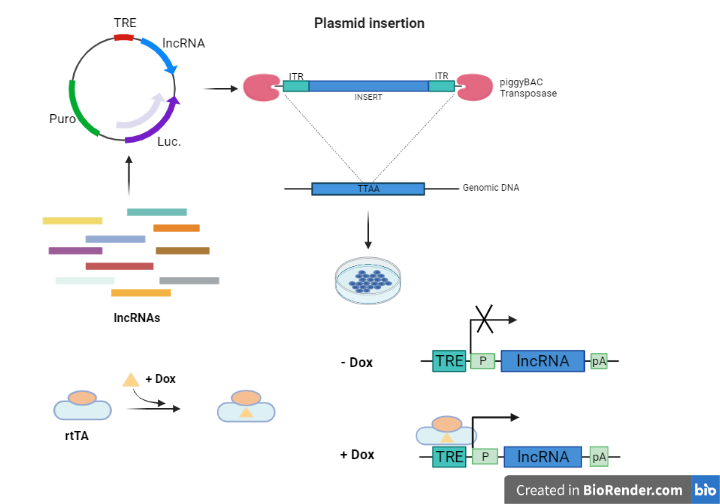
Figure 2 – Overview of Tetris assay. Created with BioRender.com
To further look into this analysis, the group investigated whether the k-mer content of a single portion of Xist-2kb could be more informative on cis-repression than the full group of 4096 Xist-2kb 6-mers. To do so, another TETRIS assay was performed, testing various deletions of Xist-2kb. This allowed to find another important sequence previously known to form secondary structure. Deletion of this region together with repA lead to full loss of the repression mechanism of Xist-2kb. After this search the authors tested a number of k-mer sets that could outperform the entire Xist-2kb set. Unfortunately, none of these k-mer sets allowed SEEKR to perform a better correlation. In the same way, changing the size of the k-mer did not result in better correlation.
In conclusion, SEEKR is presented as an innovative, well-functioning tool able to address issues that older algorithms cannot. K-mer content is proven to be a great proxy for quantifying the similarity of lncRNA sequences, which can lead to functional classification. However, the amount of data on which the algorithm has been tested is relatively small, and only cis-repression as a mechanism has been addressed; it needs to be evaluated on a larger, more various data pool to be given a more reasonable verdict.
In a future perspective, one could suggest an evolution of SEEKR in the usage of probabilistic models (position weighted matrix) in place of the k-mers. While k-mers are, in a sense, simple “words” counted inside the sequences, PWMs account for the relative probability of having a certain nucleotide in each position. It is fair to say that this process would make the complexity of the algorithm much higher, but it may also lead to new important discoveries. SEEKR might prove helpful in tandem with other approaches and/or algorithms. One example of this is the ImmLnc algorithm, which identifies immune-related lncRNAs based on their increased expression in immune cells[10]. Immune-related lncRNAs may be further characterized through their k-mer content.
References
- Kirk, J.M., Kim, S.O., Inoue, K. et al. Functional classification of long non-coding RNAs by k-mer content. Nat Genet 50, 1474–1482 (2018). https://doi.org/10.1038/s41588-018-0207-8
- Statello, L., Guo, CJ., Chen, LL. et al. Gene regulation by long non-coding RNAs and its biological functions. Nat Rev Mol Cell Biol 22, 96–118 (2021). https://doi.org/10.1038/s41580-020-00315-9
- Thompson JD, Linard B, Lecompte O, Poch O A Comprehensive Benchmark Study of Multiple Sequence Alignment Methods: Current Challenges and Future Perspectives. PLoS ONE 6(3): e18093 (2011). https://doi.org/10.1371/journal.pone.0018093
- Hezroni H, Koppstein D, Schwartz MG, Avrutin A, Bartel DP, Ulitsky I. Principles of long noncoding RNA evolution derived from direct comparison of transcriptomes in 17 species. Cell Rep.;11(7):1110-22 (2015) doi: 10.1016/j.celrep.2015.04.023.
- Wheeler, T. J. & Eddy, S. R. nhmmer: DNA homology search with profile HMMs. Bioinformatics 29, 2487–2489 (2013). doi: 10.1093/bioinformatics/btt403
- Rice, P., Longden, I. & Bleasby, A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16, 276–277 (2000) doi: 10.1016/s0168-9525(00)02024-2
- Dunham, I. et al. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57-74 (2012) https://doi.org/10.1038/nature11247.
- Van Nostrand, E. L. et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat. Methods 13, 508-514 (2016). https://doi.org/10.1038/nmeth.3810
- Lee JT, Bartolomei MS. X-inactivation, imprinting, and long noncoding RNAs in health and disease. Cell. Mar 14;152(6):1308-23 (2013). doi: 10.1016/j.cell.2013.02.016. PMID: 23498939.
- Li, Y., Jiang, T., Zhou, W. et al. Pan-cancer characterization of immune-related lncRNAs identifies potential oncogenic biomarkers. Nat Commun 11, 1000 (2020). https://doi.org/10.1038/s41467-020-14802-2.