
Relationships between DNA methylation, chromatin accessibility and gene expression studied through ATAC-Me

Abstract
ATAC-Me is a new developed approach in molecular biology which detects simultaneously chromatin accessibility and DNA methylation considering the same cell population. This technique reveals an unexpected and significant temporal disconnect between DNA methylation, chromatin accessibility, and gene expression at enhancers during monocyte-to-macrophage transitions of THP-1 cells.
Methylation of DNA (DNAme) occurs in all living organisms and most of the times the methyl group is added to cytosines, causing their conversion in 5-methylcytosines, localized in gene regulatory regions (GREs) like enhancers or promoters. This kind of methylation is a form of gene expression regulation: DNAme is typically associated with repression of promoters and enhancers, whereas low levels of DNAme usually reflect the active transcription of a gene. Chromatin accessibility (ChrAcc) plays a fundamental role in the regulation of gene expression, too. When a region of chromatin is accessible, RNA polymerase can transcribe genes in its proximity, but if chromatin does not show accessibility, gene expression is not allowed anymore. What are the relationships between DNAme and ChrAcc?
A new technique, called ATAC-Me, has been developed by Barnett and collaborators [1]. to answer this question ATAC-Me can be imagined as the combination of two other techniques: transposase-assisted enrichment of DNA fragments deriving from accessible chromatin, that allows accessible regions of chromatin to be determined [2], and bisulfite conversion (followed by deep sequencing), which helps in discriminating between unmethylated and methylated cytosines [3]. All the experiments performed by Barnett and his colleagues have a specific goal: reaching a deeper understanding of the spatiotemporal relationships between DNAme and ChrAcc. Trough ATAC-Me they collected data of DNAme and ChrAcc at myeloid enhancers at specific time points during a time course of THP-1 monocyte exposure to PMA. In parallel they performed RNA-Seq, collecting data at each time point, but also ATAC-Seq and WGBS at 0 and 24 h to have a key reference to compare data from ATAC-Me with. The cells used in this study are THP-1 monocytes, belonging to a human cell line that derives from a monocytic leukemia and, after a short exposure to PMA, differentiate in macrophages [4].
First of all, is ATAC-Me reproducible? Are ATAC-Me data obtained in accordance with ATAC-Seq data? A comparison of DNA fragments deriving from ATAC-Me and ATAC-Seq is performed: DNA fragment size range is preserved, even if a clear loss of genetic material occurs (caused by bisulfite conversion). ATAC-Me peak locations is determined genome-wide and there seem to be a correspondence with peaks estimated by previous ATAC-Seq analysis. ATAC-Me and standard ATAC-Seq performances are compared at 0 and 24 h and the result underlines a high concordance between data acquired through these two techniques.
Once had the confirmation of ATAC-Me reproducibility and its concordance with ATAC-Seq, ATAC-Me is used to measure cytosines methylation at CpG sites in DNA fragments deriving from chromatin accessible regions. DNAme levels obtained through ATAC-Me are compared with those from WGBS at 0 and 24 h within peak regions. A high concordance of data coming from these two techniques can be clearly seen. It is also relevant to highlight that ATAC-Me measures DNAme only in accessible chromatin regions. To demonstrate this, DNAme levels are measured through ATAC-Me and WGBS in some allelic regulated regions (ARRs): the former detects lower DNAme levels with respect to the latter. This happens because ATAC-Me “sees” only the accessible allele of ARRs, while WGBS takes both the alleles into account as it does not consider the real accessibility of DNA fragments in cells.
After analysing and clustering data obtained through ATAC-Me, five groups of chromatin regions with different accessibility behaviours during the time course are identified: 1) gradual closing, 2) transient, 3) early persistent, 4) gradual opening, 5) late response. Each cluster shows a differential motif enrichment of particular transcription factors (TFs). Among the TFs able to recognise one of the found TF footprints, some are known to be part of protein kinase C pathway, a protein representing a target of PMA used to stimulate THP-1 cells. These TF footprints are coherent with process of TFs binding at specific time points.
Then Barnet and his colleagues compared DNAme levels within static chromatin regions with those of dynamic. If all chromatin accessible regions (both dynamic and static) are taken into account, a prevalence of hypomethylation is observed. On the contrary, if only dynamic chromatin regions are considered, there is a significant number of peaks showing hypermethylation. In particular, heatmaps of DNAme levels, measured in the five clusters regarding different accessibility behaviours of chromatin across time, show some “contrasting” features that need to be cited: low DNAme levels in gradual closing regions even though chromatin is losing accessibility, constant DNAme levels in transient regions even though chromatin is changing through time, high DNAme levels in gradual opening regions even though chromatin is gaining accessibility. This means that DNAme levels and ChrAcc are decoupled molecular events!
At this point RNA-Seq data were collected at each time point and clustered, but only the top 20% genes whose expression was more variable during the time course is taken into consideration. What was noticed is that clusters regarding transcriptional response changes are very similar to the five clusters regarding ChrAcc behaviours across time. With that being said, transcript abundances are compared with ChrAcc and DNAme levels, always considering only the top 20% most variable genes associated with a dynamic chromatin region. A strong positive correlation links transcript abundances and ChrAcc, while there is an extremely weak relationship between transcript abundances and DNAme. This brings us to a simple conclusion: ChrAcc allows to predict transcriptional responses more efficiently than DNAme levels (Fig.1).Figure 1: standardized difference though time considering DNAme state of chromatin-accessible DNA fragments, ATAC read counts, read counts of neighbouring gene transcripts (only top 20% most variable). The plot refers to dynamic chromatin regions showing gradual opening response during the time course (0-24 h).
Maybe changes in ChrAcc and DNAme are two events occurring in two different timescales. To verify this hypothesis, chromatin regions belonging to early persistent, gradual opening and late response clusters are studied during an extended time course of PMA induction in which ATAC-Me data are collected at 24, 48, 72 h. A small loss of DNAme is observed at 72 h in these regions. Finally, THP-1 cells are treated with both PMA and vitamin C (350μM), a molecule able to enhance the activity of TET enzymes involved in DNA demethylation. Effectively, at the end of a time course of 72 h, an important loss of methylation is observed in the considered chromatin regions and this loss is much bigger if compared to that occurring in cells not treated with vitamin C.
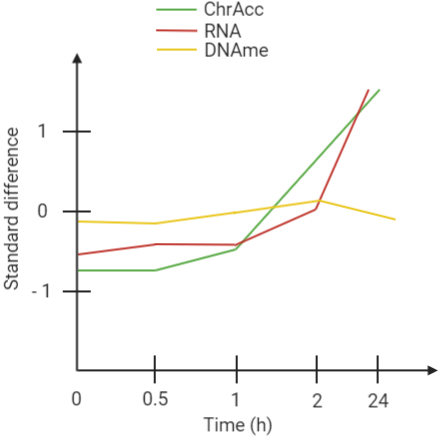
Figura 1
In conclusion, thanks to a new technique, called ATAC-Me, has been demonstrated a temporal disconnection between ChrAcc and DNAme and, the former seems to be a better predictor of transcriptional responses than DNAme levels.
References
- Barnett, K.R., Decato, B.E., Scott, T.K., Attalla, J., Smith, A.D., and Hodges, E. (2020). ATAC-Me captures prolonged DNA methylation of dynamic chromatin accessibility loci during cell fate transitions. Molecular Cell 77, 1-15.
- Buenrostro, J.D., Giresi, P.G., Zaba, L.C., Chang, H.Y., and Greenleaf, W.J. (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213-1218.
- Adey, A., and Shendure, J. (2023). Ultra-low-input, tagmentation-based whole-genome bisulfite sequencing, Genome Res. 22, 1139-1143.
- Tsuchiya, S., Kobayashi, Y., Goto, Y., Okumura, H., Nakae, S., Konno, T., and Tada, K. (1982). Induction of maturation in cultured human monocytic leukemia cells by phorbol diester. Cancer Res. 42, 1530-1536.