
Resistance mechanism of EGFR-mutant non-small cell lung cancer mediated by YAP

Figure 1. Schematic representation of the effects of three drug treatments on EGFR-mutant NSCLC cells.
Abstract
Abstract
The treatment of EGFR-mutant non-small cell lung cancer (NSCLC) cells with the combination of epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) and MEK inhibitor induces the development of a senescence-like dormant state. In a recent publication Kurppa et al. [1] demonstrate that the dormant state is characterized by an increase in YAP/TEAD activity that is responsible for the survival of NSCLC cells to apoptosis induced by combined EGFR/ MEK inhibition. YAP, TEAD and SLUG form a complex that suppresses drug-induced apoptosis through transcriptional repression of pro-apoptotic protein BMF. YAP/TEAD inhibition enhances EGFR/MEK inhibition-induced apoptosis and causes a reduction in the population of dormant therapy-resistant cancer cells.
Discussion
Genotype-directed therapy is the standard therapy for many cancers that harbor an activated oncogene, however this kind of therapy is rarely capable of completely eradicate a tumor. This is due to the fact that, after drug treatment, the residual cancer cells can serve as reservoirs for the development of acquired drug resistance [2]. Patients affected by EGFR-mutant NSCLC usually are treated with Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) that are able to block the excessive proliferation of cells [3]. However, EGFR-mutant cancer cells, after the treatment with EGFR TKI, can enter a drug-tolerant state; in this way cancer cells are able to evade apoptosis and survive. Consequently, due to the emergence of acquired resistance there is a reduction of the clinical effectiveness of the treatment [4]. A short time after the EGFR TKI treatment, despite the inactivation of EGFR, there is the reactivation of ERK1/2 a molecule that in EGFR signalling pathway is located downstream from EGFR [5]. The combination of EGFR inhibitor with a MEK inhibitor is able to prevent the reactivation of ERK1/2 and there is an increase in the apoptotic response of cancer cells compared to single-agent EGFR inhibition. However, even with this combination, acquired resistance can still develop [6]. The aim of the study of Kurppa et al. [1] is to understand the mechanisms that allow cancer cells to evade apoptosis and survive despite combined EGFR/MEK inhibition.
A study of the enrichment of transcription factor binding sites and immunofluorescence staining allowed Kurppa et al. to observe that in the cancer cells treated with EGFR/MEK inhibitors, compared to control cells, there was an increase in the activity of transcriptional factor TEAD and its partner YAP. By loss of function experiments Kurppa et al. demonstrated that the hyperactivation of YAP/TEAD is an important regulatory mechanism in EGFR-mutant NCSLC that is able to promote cell survival and establish a drug-resistant non-proliferative state reducing in this way the effectiveness of the treatment with EGFR/MEK inhibitors. By RNA-seq experiment the authors discovered that YAP/TEAD are able to mediate cell survival by repressing the expression of BMF gene which encodes for a pro-apoptotic protein. They also demonstrated that YAP and TEAD don’t work alone but they are able to cooperate with SLUG, a transcription factor mediating Epithelial mesenchymal transition (EMT). These three transcription factors form a tertiary complex that is responsible for the transcriptional repression of BMF following EGFR/MEK treatment and in this way the tertiary complex limits the initial drug-induced apoptotic effect allowing the formation of a dormant state.
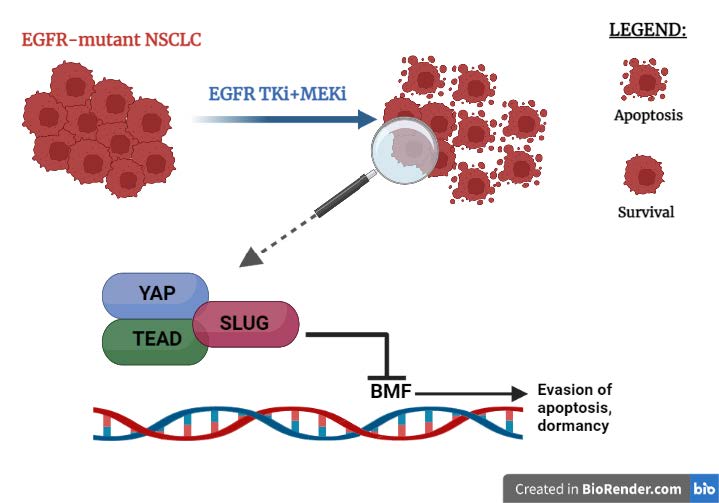
Figure 2. Some EGFR-mutant NSCLC cells survive the combined treatment of EGFR/MEK inhibitors thanks to the YAP/TEAD/SLUG complex binding DNA and preventing the expression of the pro-apoptotic gene BMF.
Thanks to an RNA-seq experiment and gene set enrichment analysis they could observe that gene expression of the dormant state is similar to gene expression of cellular senescence as the dormant state shares several characteristics of cellular senescence. Using live-cell imaging and western blot analysis they observed that EGFR-mutant NSCLC cells adopt only temporarily the dormant state induced by the activation of YAP as long as the EGFR/MEK inhibitors are present in the environment in order to be able to tolerate the lethal drug exposure. Following drugs withdrawal, cancer cells exit the senescence-like dormant state and they begin to proliferate again. Consequently, the senescent-like population can serve as a reservoir of dormant cells that, after the removal of the drug, are able of proliferate and re-establish the tumor. Thus, demonstrating that the increase of YAP/TEAD activity is essential for the establishment of the resistant state upon treatment; consequently, YAP and TEAD are attractive targets for drug development.
Flufenamic acid is a molecule that can bind TEAD and using this molecule as a structural basis the authors of the study managed to develop a novel covalent TEAD inhibitor, called MYF-01- 37, that is able to prevent the interaction between YAP and TEAD. The authors performed a qPCR-experiment in vitro to demonstrate its effectiveness. The study suggests that the combination of EGFR/MEK inhibitors with YAP/TEAD inhibitors, such as MYF-01-37 or XAV939, has great potential to increase the effectiveness of the treatment in EGFR-mutant NSCLC and limit the establishment of the resistant state.
It is unknown if the suppression of apoptosis through transcriptional repression of BMF mediated by YAP is a resistance mechanism that, aside from EGFR-mutant NSCLC, is also present in other kind of cancers.
However, to allow MYF-01- 37 to undergo preclinical testing using in vivo models of EGFR-mutant NSCLC it is necessary to further optimize its pharmacological properties. This treatment strategy will need to undergo clinical trial to assess both its efficacy and its toxicity.
Conclusions
In this study Kurpa et al. discovered a new mechanism that cancer cells use to resist to clinical treatment, making an important contribution to the research of new therapies against EGFR-mutant NSCLC.
The authors concluded that the treatment with EGFR/MEK inhibitors is responsible for the induction of the dormant state, but it can’t be excluded with confidence the possibility that the dormant cells exist already in the initial population and that the treatment with inhibitors kills all the other cells uncovering this subpopulation. Besides, even if the combination of EGFR/MEK inhibitors with YAP/TEAD inhibitors induces a robust apoptotic response in cancer cells, this combination is unable to completely eradicate the tumor but it is only able to slow down his growth compared to EGFR/MEK inhibitors alone. In fact, cannot be excluded that, in addition to YAP/TEAD/SLUG, there are other factors
involved in the long-term survival of EGFR-mutant cancer cells treated with EGFR/MEK inhibitors. Future studies will have to clarify these doubts.
References
- Kurppa et al., 2020, Cancer Cell 37, 104–122 January 13, 2020 2019 Elsevier Inc.
- Drilon, A. et al. (2018). Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N. Engl. J. Med. 378, 731–739.
- Mok, T.S. et al. (2009). Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 361, 947–957.
- Cortot, A.B., and Janne, P.A. (2014). Molecular mechanisms of resistance in epidermal growth factor receptor-mutant lung adenocarcinomas. Eur. Respir. Rev. 23, 356–366.
- Ercan, D. et al. (2012). Reactivation of ERK signaling causes resistance to EGFR kinase inhibitors. Cancer Discov. 2, 934–947.
- Tricker, E.M. et al. (2015). Combined EGFR/MEK inhibition prevents the emergence of resistance in EGFR-mutant lung cancer. Cancer Discov. 5, 960–971.