
Slide-DNA-seq: a novel technique to study cancer heterogeneity

Picture 1 – Schematic representation of the slide-DNA-seq analysis workflow
Abstract
Tumoral tissues are generally highly heterogeneous, displaying diverse genetic aberrations. Golden standards already present in the field are not able to study aberrations without the loss of spatial distribution information. Here we show slide-DNA-seq, a technique that enables spatially resolved DNA sequencing from intact tissues. Slide-DNA-seq was able to detect cancer cells within a healthy tissue and to discriminate between different tumoral subclones. We anticipate slide-DNA-seq to become a valid alternative to the laser-capture microdissection and single-cell DNA sequencing methods for clinical diagnosis.
Discussion
In the tumor, each of the cancer cells is able to evolve a different repertoire of DNA mutations, copy number alterations (CNAs) and large chromosomal rearrangements which give rise to different clonal populations within the tumor 1. Having the ability to study which kind of mutations the tumor cells have acquired and how they are arranged in the tumor is of paramount importance because the tumor environment defines some properties of the tumor itself such as the ability to resist drugs, the ability to resist immune cells or the ability to metastasize 2.
Current methods to detect cancer heterogeneity includes deep sequencing to quantify mutant allele frequencies and single-cell whole-genome sequencing (scWG-seq). Notably, these methods can detect genetic alterations that occurs during the evolution of the tumor but do not measure spatial organization of the cancer cells within the tumor and the healthy tissue3.
The golden standards to measure spatial organization currently are Laser Capture Microdissection (LCM) and Hematoxylin and Eosin (H&E) staining 4. Both are constrained to capture and analyze late stage tumoral masses, which have to be picked manually (LCM) or seen (H&E) by a skilled operator, limiting de-novo discovery. The need for a bias-less technique to discover and discriminate clonal population within the tumor is then, of great relevance.
A solution comes from a recent publication of Zhao et al. 5 from Harvard University with slide-DNA-seq, which allows spatial resolved DNA sequencing from intact tissues.
First, a spatially indexed array of 10 µm polystyrene beads ligated with an unique barcode needs to be generated. Following tissue cryosection, a single 10 µm thick frozen section is transferred onto the sequenced bead array. Genomic fragments are generated with TN5 transposase, an enzyme which is able to cut and in the meanwhile insert custom adapter sequences: a bead adapter and an Illumina adapter. Subsequently, hybridization of a bridge adapter enables ligation of photocleaved, spatially indexed bead oligonucleotides to proximal genomic fragments. The resulting DNA sequences are amplified by PCR to obtain a DNA sequencing library, and then sequenced. The barcodes were used to reconstruct the spatial organization of DNA in the tissue.
Picture 1 – Schematic representation of the slide-DNA-seq analysis workflow
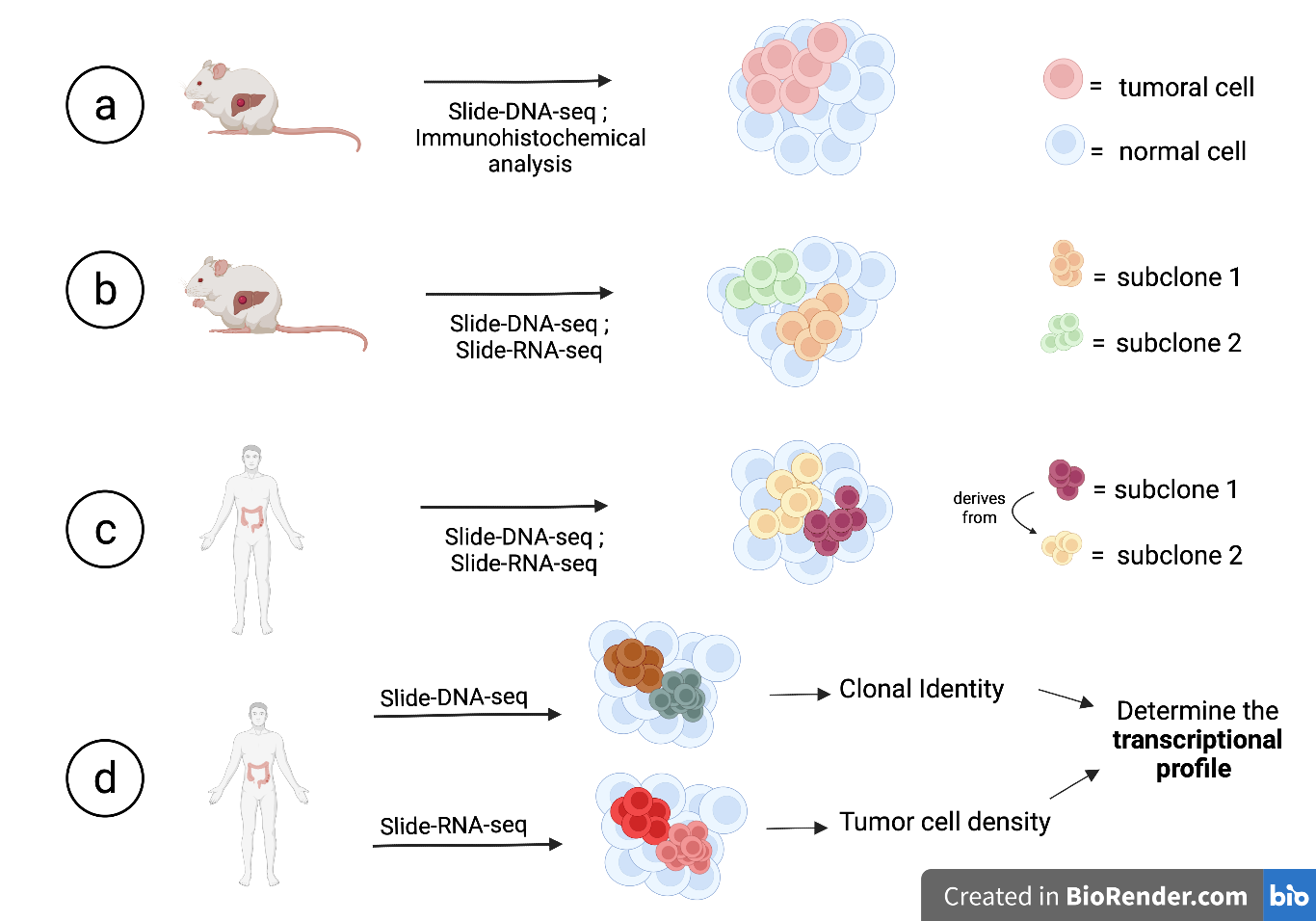
The first objective of the authors was to discriminate healthy and tumoral tissues by applying slide-DNA-seq to mouse liver tissue slides displaying metastases (Fig.2A). After sequencing, they performed PCA and k-means clustering in order to divide the cells into either healthy or tumoral. The results were concordant with an immunohistochemical experiment performed by using the tumoral marker HMGA2.
Another experiment was performed in order to demonstrate if the technique was also able to discriminate between different clonal populations within the same tumor (Fig.2B). They took a tissue slide in which H&E staining revealed two distinct tumoral masses and, performing slide-DNA-seq, different chromosomal aberrations were found, driving the two tumoral masses to be divided and recognized as different clones. Slide-RNA-seq [6] was also performed on the same tissue slide, confirming that the previously seen clones were also transcriptionally different suggesting that they took divergent paths in their tumoral evolution and a functional heterogeneity.
Further, they applied slide-DNA-seq to a human stage III colorectal cancer in order to see if clones within the tissues derived from a single tumoral ancestor or from different ones (Fig.2C). Slide-DNA-seq performed on a tissue slide divided the cells into two tumoral subclusters and a healthy one. All the cells recognized as tumoral displayed variance under PC1, with only a subpart of them displaying variance for PC2, suggesting tumoral evolution of one subclone from the other. Integration with scWG-seq on the same slide, resulted in the recognition of additional tumoral subclones due to higher resolution. This also provides a temporal information about the progress of tumoral evolution and demonstrates the utility of slide-DNA-seq as a tool to study clonal heterogeneity.
The next step was to find out whether tumor transcriptional programs were controlled by genetics and the environment surrounding the tumor. They took a different tissue slide from another colorectal cancer and performed slide-DNA-seq along with slide-RNA-seq (Fig.2D). Slide-DNA-seq divided the cells into different subclones and slide-RNA-seq was used to quantify for tumoral cell density. Differentially expressed genes associated with the different subclones or with high tumoral density were then used to perform a gene set enrichment analysis. They found that the genes driving for the division into the clusters were mainly involved in tumor progression, while genes driving for higher or lower cell density signals were genes coding for cell adhesion molecules and cadherins.
Conclusions
This study provides the proof that slide-DNA-seq is able to discriminate between healthy and cancer tissues, detect clonal heterogeneity and most importantly, preserve spatial distribution information. Even if slide-DNA-seq was enough to satisfy all the previous tasks, the integration with other techniques (slide-RNA-seq, scWG-seq) improved detection of divergent paths and provided functional information on the tumor biology. It is worth to note that this kind of approach would be hardly appliable as golden standard in clinical diagnosis and prognosis.
Slide-DNA-seq proved itself to be a viable diagnostic tool to detect de-novo clonal populations within tumors with relative ease. The technique itself, based mainly on sequencing could be applied to other, of great relevance as well, scientific fields such as:
- creating a “tumoral evolution atlas”
- studying the differentiation of normal, healthy tissues
- special evaluation of other aspects of genomics, like DNA-methylation, modifying slightly the technique
The implementation of slide-DNA-seq as a potential future golden standard for clinical diagnosis, remains, though, as of now a hopeful future perspective. We cannot expect all medical infrastructures in the world to have this cutting-edge sequencing technology and the qualified personnel to perform the needed experiments. Standard H&E staining analysis of potential cancer tissues remains as of now easier and most importantly cheaper.
References
- McGranahan, N. & Swanton, C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell 168, 613-628 (2017).
- Pogrebniak, K. L. & Curtis, C. Harnessing tumor evolution to circumvent resistance. Trends Genet. 34, 639-651 (2018).
- Gerstung, M. et al. The evolutionary history of 2,658 cancers. Nature 578, 122-128 (2020).
- Casasent, A. K. et al. Multiclonal invasion in breast tumors identified by topographic single cell sequencing. Cell 172, 205–217.e12 (2018).
- Zhao T, Chiang ZD, Morriss JW, LaFave LM, Murray EM, Del Priore I, Meli K, Lareau CA, Nadaf NM, Li J, Earl AS, Macosko EZ, Jacks T, Buenrostro JD, Chen F. Spatial genomics enables multi-modal study of clonal heterogeneity in tissues. Nature. 2022 Jan;601(7891):85-91. doi: 10.1038/s41586-021-04217-4. Epub 2021 Dec 15. PMID: 34912115.
- Stickels RR, Murray E, Kumar P, Li J, Marshall JL, Di Bella DJ, Arlotta P, Macosko EZ, Chen F. Highly sensitive spatial transcriptomics at near-cellular resolution with Slide-seqV2. Nat Biotechnol. 2021 Mar;39(3):313-319. doi: 10.1038/s41587-020-0739-1. Epub 2020 Dec 7. PMID: 33288904; PMCID: PMC8606189.